Rétinite pigmentaire
Survol
La rétinite pigmentaire (RP) désigne un groupe d’affections génétiques qui portent atteinte aux cellules photosensibles de la rétine et entraînent une perte de vision graduelle, à mesure que ces cellules meurent. Considérée comme une « maladie rare », la RP est pourtant l’une des maladies héréditaires de la rétine les plus courantes, touchant entre 1 Canadien sur 3 500 à 4 000[1]. La RP est souvent appelée maladie héréditaire de la rétine, ce qui signifie qu’elle est transmise entre lignées génétiques et qu’une personne la reçoit par hérédité de ses parents. Généralement, la rétinite pigmentaire est diagnostiquée à l’enfance ou à l’adolescence, mais une minorité de patients ne présentent aucun symptôme avant l’âge adulte.
Les cellules spécialisées appelées photorécepteurs sont responsables de capter la lumière et de la traduire en signaux que le cerveau interprétera; ce sont ces cellules-là qui meurent peu à peu chez la personne atteinte de RP. Les photorécepteurs sont de deux types : les bâtonnets rétiniens et les cônes rétiniens. Les bâtonnets sont responsables de la vision périphérique et nocturne, tandis que les cônes rétiniens sont responsables de la vision centrale et de la perception des menus détails et des couleurs. Comme la RP porte d’abord atteinte aux bâtonnets rétiniens, la perte de vision périphérique et nocturne apparaît dès les premiers stades de la maladie, après quoi le champ de vision se rétrécit, ce que l’on appelle souvent la forme progressive de « vision tubulaire ». Les bâtonnets rétiniens continuent de disparaître et, avec le temps, les cônes rétiniens commencent à faire de même. La disparition des cônes rétiniens entraîne la perte de la vision centrale et mène bien souvent, aux derniers stades de la maladie, à la cécité totale ou quasi totale. La durée de ce processus varie d’une personne à l’autre.
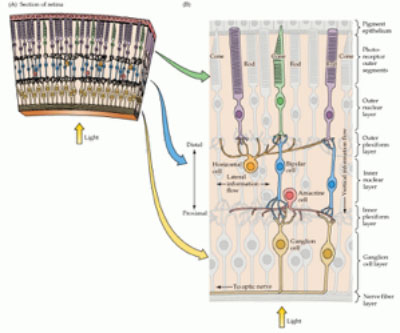
La RP était considérée à l’origine comme une seule maladie, mais après des dizaines d’années de recherche, notamment la recherche financée par VCC, nous savons maintenant qu’il existe plusieurs types de RP et que ces derniers impliquent des mutations dans un gène parmi plus de 64 gènes différents. Le type de la maladie et ses symptômes dépendent ainsi du ou des gènes ayant muté.
Il y a divers modes de transmission héréditaire de la RP, ce que l’on appelle généralement le « profil de transmission héréditaire » : autosomique dominant, autosomique récessif ou récessif lié au chromosome X. Un conseiller en génétique pourra discuter avec vous de vos antécédents familiaux afin de déterminer lequel de ces modes est associé à votre perte de vision. Ces informations pourraient aussi lui permettre de vous en dire davantage sur la progression de votre maladie et de vous renseigner sur le risque de perte de vision chez les membres de votre famille. Pour en savoir plus sur le dépistage génétique de la RP, veuillez lire le texte Dépistage génétique rédigé par VCC.
En règne générale, chez une personne atteinte de RP, une seule paire de gènes présente des anomalies. Les scientifiques ont à ce jour ciblé plus de 64 gènes où des mutations qui causent la RP peuvent se produire, et on finira probablement par cerner des mutations dans plus de 100 gènes différents. Puisqu’un si grand nombre de mutations génétiques à l’origine de la RP demeurent inconnues, les chances qu’un dépistage génétique donne des résultats définitifs sont d’environ 50 %. En se basant sur vos antécédents familiaux et sur le mode de transmission héréditaire de la RP dans votre cas, le conseiller en génétique pourra vous dire si les résultats du dépistage génétique sont susceptibles d’êtres définitifs.
Diverses mutations génétiques peuvent endommager la rétine ou entraver son fonctionnement de différentes façons. Par exemple, certaines mutations modifient la façon dont la rétine transforme les nutriments, alors que d’autres portent atteinte aux photorécepteurs. Il importe de déterminer le gène et la mutation responsables de la maladie, car de nombreux traitements contre la RP en cours de développement cibleront des types génétiques spécifiques.
Le contenu de cette page a été rédigé par les Drs Chad Andrews et Mary Sunderland. Il été mis à jour le 23 août 2018. Une version antérieure avait été approuvée par les Drs Jane Green et Bill Stell.
Unissons nos forces
Découvrez comment votre soutien peut contribuer à tracer un avenir sans cécité! Soyez informé en exclusivité des dernières percées de la recherche en santé de la vision et des événements dans votre région en vous abonnant à notre infolettre.